Nov 3 2012
A University of Cincinnati (UC) cancer biology team reports breakthrough findings about specific cellular mechanisms that may help overcome endocrine (hormone) therapy-resistance in patients with estrogen-positive breast cancers, combating a widespread problem in effective medical management of the disease.
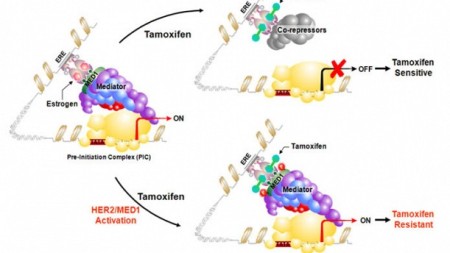 Zhang's research model showing HER2 activation of MED1 drives estrogen receptor corepressor/coactivator switch by tamoxifen
Zhang's research model showing HER2 activation of MED1 drives estrogen receptor corepressor/coactivator switch by tamoxifen
Xiaoting Zhang, PhD, and his colleagues have identified a specific estrogen receptor co-activator—known as MED1—as playing a central role in mediating tamoxifen resistance in human breast cancer. The team reports its findings in the Nov. 1, 2012, issue of Cancer Research, a scientific journal of the American Association for Cancer Research.
According to the National Cancer Institute, nearly 227,000 women are diagnosed with breast cancer annually in the United States. About 75 percent have estrogen-positive tumors and require adjuvant hormone therapy such as tamoxifen, a drug that works by interfering with estrogen’s ability to stimulate breast cancer cell growth.
Despite advances in hormone therapy drugs, cancer surveillance research has shown that 50 percent of patients will develop resistance to the drug and experience a cancer relapse.
The hormones estrogen and progesterone can stimulate the growth of some breast cancers. Hormone therapy is used to stop or slow the growth of these tumors. Hormone-sensitive (i.e., positive) breast cancer cells contain specific proteins known as hormone receptors that become activated once hormones bind to them, leading to cancer growth.
Based on new findings, UC Cancer Institute scientists believe that tamoxifen resistance may be driven by a novel molecular "crosstalk” point between the estrogen and HER2 (human epidermal growth factor receptor 2) signaling pathways.
Testing in both pre-clinical models and human breast cancer tissue samples showed that MED1 co-amplifies and co-expresses with HER2, a gene that has an increased presence in 20-30 percent of invasive human breast cancer and plays a major role in tamoxifen resistance.
HER2 over-expression led to MED1 activation while reduction of MED1 caused breast cancer cells that were otherwise tamoxifen-resistant to respond and stop dividing. Further mechanistic studies showed that HER2 activation of MED1 resulted in the recruitment of co-activators instead of co-repressors by tamoxifen-bound estrogen receptor. This, explains Zhang, drives expression of not only traditional estrogen receptor-positive cancer target genes, but also HER2 and those estrogen receptor target genes abnormally activated by HER2.
"Together, these findings suggest this ‘crosstalk’ could play a central role in mediating tamoxifen resistance in human breast cancer, especially because recent published data also indicated that high MED1 expression levels correlate with poor treatment outcome and disease-free survival of patients who underwent endocrine therapy,” explains Zhang, an assistant professor of cancer biology at the UC College of Medicine and breast cancer researcher with the UC Cancer Institute.
"We are currently utilizing RNA-based nanotechnology to target MED1 in an effort to simultaneously block both estrogen and HER2 pathways to overcome endocrine-resistant breast cancer.”
UC study collaborators include cancer biologists Jiajun Cui, PhD, Katherine Germer, MD, Shao-chun Wang, PhD; environmental health researcher Tianying Wu, PhD; and pathologist Jiang Wang, MD. Qianben Wang, PhD of the Ohio State University College of Medicine, and Jia Luo, PhD, of the University of Kentucky, also contributed to this study.
The study was supported with start-up funding from the UC Cancer Institute, Ride Cincinnati/Marlene Harris Pilot Grant, Susan G. Komen for the Cure Foundation and the Center for Clinical and Translational Science and Training—home to UC’s institutional Clinical and Translational Science Award program grant from the National Institutes of Health.
Source: http://www.uc.edu/