The mass production of pharmaceuticals has significantly evolved since the industrial fabrication of synthetic drugs towards the end of the 19th century1 with efficient developments in process control, sterility and technology guidelines.
Even though optimizations in pharmaceuticals are continually developing, recalls still regularly happen2 and their severe impact on both the consumer and the manufacturer are the reasons why international regulation of the sector is so strict.
Environmental monitoring (EM) is one element of the wider landscape of current good manufacturing practices (cGMPs) that has been standardized for the protection of human health.
EM is enforced through the evaluation of viable and non-viable particles to uphold a dependable degree of control over production in a standard cleanroom.
The European Commission’s EU GMP Annex 1 2020 Draft and the Sterile Drug Products Produced by Aseptic Processing from the FDA both define the restrictions in relation to an area’s grade or classification.

Nonviable monitoring with the Airnet® II aerosol particle sensor (left), viable monitoring with the MiniCapt® remote microbial impactor (right). Image Credit: Particle Measuring Systems
Viable particles are living organisms like yeasts, bacteria and molds. The EU GMP guidelines can be viewed in ‘Table 7, Maximum action limits for viable particle contamination’.3 The FDA cGMP standards are presented in ‘Table 1, Air classifications’.4
Non-viable particles function as transportation for viable particles and do not include a living organism. The EU GMP standards can be read in, ‘Table 6, Limits for airborne particulate concentration for the monitoring of non-viable contamination’.3 FDA cGMP standards can be viewed in ‘Table 1, Air classifications’.4
Historically, sampling techniques for the quantification of potential contamination have been the main focus, such as active air samplers, particle counters, swabs for surface monitoring and Petri dishes.
The integrity of the filter and the bioburden of bulk product were also measured to support the identification of microbiological contamination throughout the stages of manufacturing.
This approach has been repurposed to manage and prevent possible contamination from infiltrating the product, which is impossible to solve.
There is no low-cost removal strategy to continue manufacturing once contamination has happened, which makes it crucial to identify the sources. This is attained by:
- Utilizing quality risk management to determine the critical control points (CCPs).
- With the CCPs, determining the stages in the process where it is crucial to establish mitigation tasks to manage the risks identified.
- Implementing Quality by Design into the area of production.
The contamination control strategy (CCS) is the term for the collective deployment of these actions.
Defining the CCS
The document, ‘EU GMP Annex 1: Manufacture of Sterile Medicinal Products’, explains what regulatory bodies recommend to be included in a CCS, with the following context:
“Contamination Control Strategy (CCS) – A planned set of controls for microorganisms, pyrogens and particulates, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in process controls, finished product specifications, and the associated methods and frequency of monitoring and control ... A Contamination Control Strategy (CCS) should be implemented across the facility in order to define all critical control points and assess the effectiveness of all the controls (design, procedural, technical and organizational) and monitoring measures employed to manage risks associated with contamination. The CCS should be actively updated and should drive continuous improvement of the manufacturing and control methods.3”
It should be noted that ‘critical control points’ do not have any related parameters and are not an In Process Control (IPC). They are instead stages in the process where a risk has been identified and must be controlled.
Finding Root Causes of Contamination
Quality Risk Management (QRM) and Quality by Design (QbD) are both innately connected to knowledge of the process. Figure 1 outlines the stages to perform a complete risk assessment of a process.
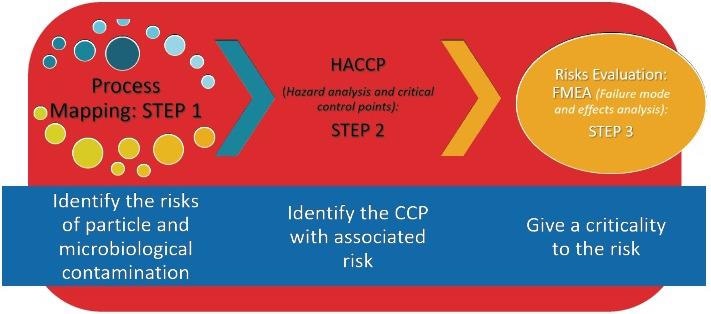
Figure 1. The steps to understanding your process. Image Credit: Particle Measuring Systems
The risks of particle ingress can be established through process mapping. A fishbone model is firstly made to record all potential contamination sources.
In Figure 2, the root sources are gathered into representative categories (Materials, personnel, environmental, methodology and equipment), but the fishbone model may require further categories to include the specific features of any given process.
As there is no precise standard to follow, knowledge of the process is critical.
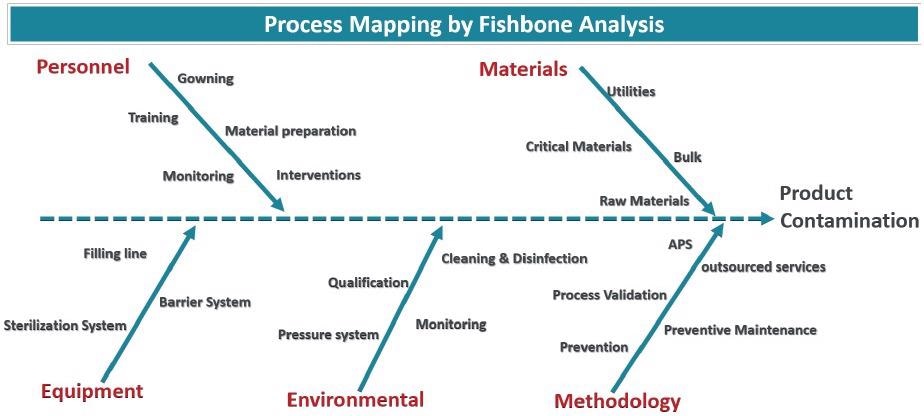
Figure 2. Fishbone diagram showcasing all sources of possible particle contamination. Image Credit: Particle Measuring Systems
Fishbone diagrams can include granularity. For example, the section of utilities within the category of materials. In this feature of the process, there are several stages that may result in contamination. An outline of the Water for Injection (WFI) stage inside the Utilities category could be presented as follows:
- Production: Downstream of WFI
- Production: After the storage tank
- Upstream of delivery pumps for the loop
- Water distribution within the loop
- Usage points
- Points of sampling
Risks to each singular input should be established after the process mapping has been completed. More than one risk may be involved with certain inputs. In line with the WFI example, this evaluation could look like the below:

Figure 3. Identifying and assigning risks to a process. Image Credit: Particle Measuring Systems
A decision tree tailored to the process is produced to establish if the risks are crucial and must be managed.
Questions will be customized at this stage to better allocate the label of CCP, which shows management is necessary to reduce contamination. There are again no fixed guidelines of yes or no questions to utilize so an understanding of the process is critical.
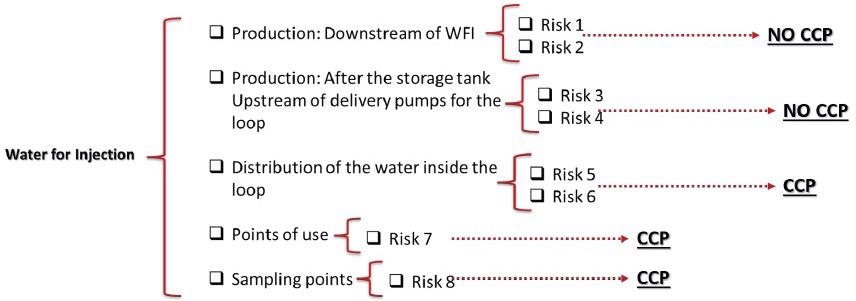
Figure 4. Determining whether a risk is critical enough to warrant control. Image Credit: Particle Measuring Systems
The HACCP technique for determining risk is qualitative, which means the risk is defined, but the actual impact on the product and the related degree of importance is not established.
The Failure Modes and Effects Analysis (FMEA) method can be employed to make the determination of risk quantitative.
Presented in Figure 5, the FMEA matrix includes a cross of three measurements. These parameters are detectability, occurrence and severity, which allows the importance of a risk to the process to be determined.
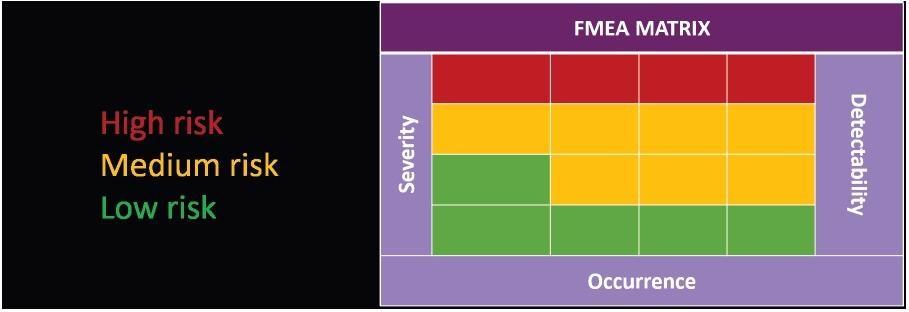
Figure 5. FMEA matrix for visualizing risk criticality. Image Credit: Particle Measuring Systems
Using this data, a mitigation and control program can be created based on the degree of importance allocated. Figure 6 demonstrates the evaluation of CCPs and their determined risk criticality.
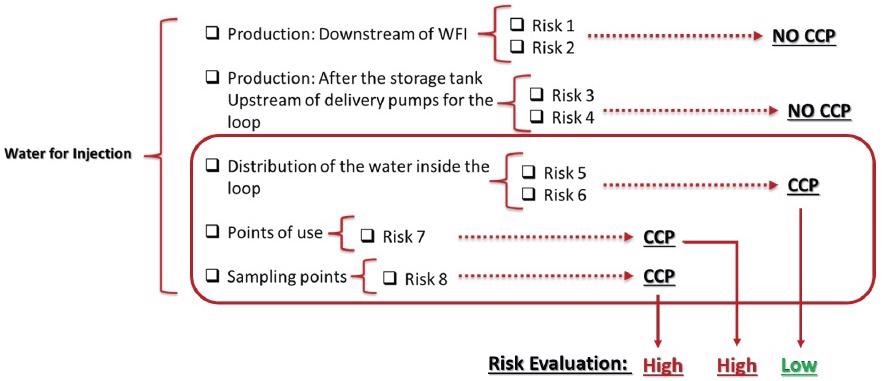
Figure 6. High and low risks from CCPs found. Image Credit: Particle Measuring Systems
Designing the Cleanroom
A risk mitigation system (Step 4 of Figure 7) including fixed controls and actions should be deployed to design a cleanroom in line with sterility assurance guidelines.
This plan could take the form of a design loop, redesign or a sequence of valves to decrease contamination. Further options can incorporate developments to sampling processes or a system re-validation or validation.
Recommended actions for risk mitigation may involve decreasing or increasing the chemical or microbiological attributes, and altering the set of controls in relation to the importance of points and processes.
Many departments, old and new, may be influenced by the execution of the mitigation plan. Due to this, the definition of the change control plan is required to organize the measures to be taken by the time required and priority.
This intervention occurs in an early stage for new cleanrooms to remove the risk of a build-up of issues that cannot be resolved further down the line.
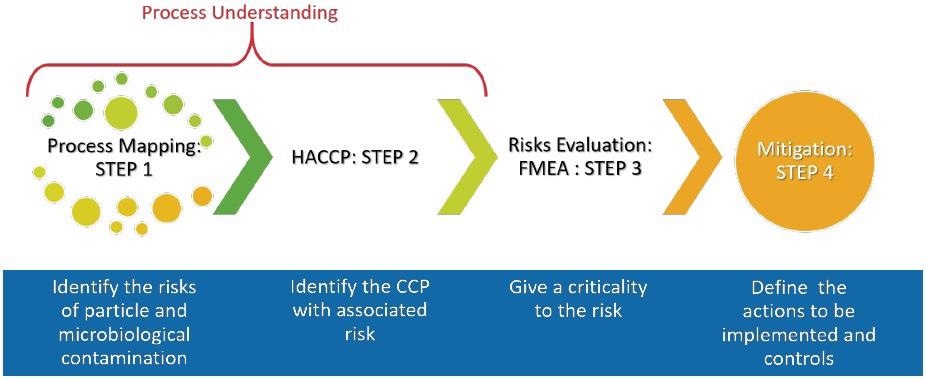
Figure 7. The final step: Risk mitigation. Image Credit: Particle Measuring Systems
The crucial element of contamination control is building a team of professionals specializing in production, quality, QRM and engineering.
The resources available at the facility should be utilized to create a trustworthy foundation of process knowledge, the QRM experts at Particle Measuring Systems can also help its customers in the creation of a contamination control strategy.
Acknowledgments
Produced from materials originally authored by Anna Campanella, PhD from Particle Measuring Systems.
References and Further Reading
- Daemmrich A, Bowden ME. The top pharmaceuticals that changed the world. A Rising Drug Industry. Chemical & Engineering News. June, 20 2005. 83(25).
- https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts. Accessed April 2, 2021.
- European Commission. EU GMP Annex 1: Manufacture of sterile medicinal products. Annex 1 Draft. Published December 2020. https://www.gmp-compliance.org/files/guidemgr/2020_annex1ps_sterile_medicinal_products_en.pdf . Accessed March 30, 2021.
- Food and Drug Administration. Guidance for Industry Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice. Published September 2004. https://www.fda.gov/. Accessed March 30, 2021.

This information has been sourced, reviewed and adapted from materials provided by Particle Measuring Systems.
For more information on this source, please visit Particle Measuring Systems.