Evidence is mounting that the development and spread of cancer, long attributed to gene expression and chemical signaling gone awry, involves a biomechanical component as well. Researchers at Lawrence Berkeley National Laboratory (Berkeley Lab) have added to this body of evidence by demonstrating that the malignant activity of a critical cellular protein system can arise from what essentially are protein traffic jams.
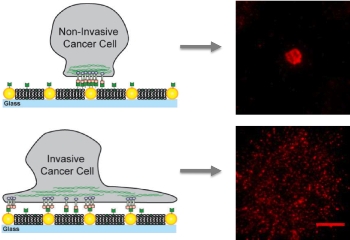 On artificial membranes embedded with gold nanodots, non-invasive cancer cells bind only to the nanodots and become immobilized while invasive cells bind to the membrane as well as the nanodots creating mobile clusters that contribute to metastasis.
On artificial membranes embedded with gold nanodots, non-invasive cancer cells bind only to the nanodots and become immobilized while invasive cells bind to the membrane as well as the nanodots creating mobile clusters that contribute to metastasis.
Using a unique artificial membrane imbued with an obstacle course of gold nanodots, a research team led by chemist Jay Groves studied the transport of the protein signaling complex EphA2/ephrin-A1 across the surfaces of 10 different breast epithelial cancer cell lines displaying a wide range of disease characteristics. The researchers found that transport of this receptor-ligand complex was normal in healthier cell lines but became jammed in diseased cell lines, with the worst jamming taking place in the cells that were the most diseased.
“There is something about how strongly the EphA2 proteins are clustered together on the cell surface that is related to and can even contribute to the malignant behavior of cancer cells,” Groves says. “The differential jamming of EphA2 transport among the various breast cancer cell lines and its correlation with disease characteristics suggests that EphA2 clustering itself may contribute to pathological effects.”
Groves holds joint appointments with Berkeley Lab’s Physical Biosciences Division and UC Berkeley’s Chemistry Department, and is also a Howard Hughes Medical Institute (HHMI) investigator. He is the corresponding author of a paper describing this research in the journal NANO Letters titled “Nanoscale Obstacle Arrays Frustrate Transport of EphA2/ephrin-A1 Clusters in Cancer Cell Lines.” Co-authors of this paper were Theobald Lohmuller and Qian Xu.
EphA2 belongs to a family of enzymes that are key regulators of cellular processes. Ephrin-A1 is a signaling protein that binds to EphA2. Complexes of EphA2/ephrin-A1 will gather in clusters that are then transported across the cell surface. The over-expression of EphA2 has been linked to a number of human cancers, but is especially prominent in breast cancer.
“Some 40-percent of all breast cancer patients show an over-expression of EphA2 that is correlated with tumor metastasis, consequently, much effort has been directed to the development of therapeutics that target EphA2,” Groves says. “However, precisely what goes wrong with EphA2 that contributes to pathological cell behavior remains unclear as EphA2 is usually not mutated in cancerous cells.”
Groves is a leading authority in the emerging field of mechanobiology, which seeks to understand how cells sense and respond to mechanical forces. To investigate for a possible mechanical factor in EphA2’s link to breast cancer, Groves used a technique his group developed in which artificial membranes made up of a fluid bilayer of lipid molecules are embedded with fixed arrays of gold nanodots. This allows researchers to control the spacing or transport of proteins and other cellular molecules placed on the membranes.
For this study, Groves and his colleagues used arrays of gold nanodots to present defined obstacles to the movement and assembly of EphA2/ephrin-A1 clusters. The ephrin-A1 ligands could bind to the membrane, which allowed the clusters to be mobile, or to the nanodots, which immobilized the clusters, or to both. The researchers worked with lines of breast cancer cells that have similar levels of EphA2 expression and included MDA-MB-231, a highly invasive and tumorigenic line, and MCF10A, a relatively benign and non-tumorigenic line.
“When we see cells that have the same levels of EphA2 but the MDA-MB-231 is jammed while the MCF10A is not, then we can say it is something beyond just the numbers of EphA2 that matters, something about the way EphA2 is plugged into the rest of the cell that is misrelated,” Groves says. “Our observations suggest the cytoskeleton is the culprit and that drugs modulating the cytoskeleton might also therapeutically modulate EphA2 clustering, thereby reducing pathological behavior.”
This research was primarily supported by the National Cancer Institute.
Source: http://www.lbl.gov/