Since the determination of the molecular structure of myoglobin in 1957, X-ray crystallography has been the defining tool of structural biology, allowing researchers to determine the structure (and hence the function) of tens of thousands of proteins, nucleic acids, and other biological molecules. But the method can only work for molecules that form large, high quality crystals.
An international team of scientists has now used the intense beam of Stanford's Linac Coherent Light Source (LCLS) [the world's first hard x-ray free-electron laser (XFEL) and one of only two currently operating in the world] to demonstrate that it is possible to reconstruct the 3D structure of a virus particle without the need of a crystal, by combining many diffraction patterns collected from a sequence of randomly oriented single viruses.
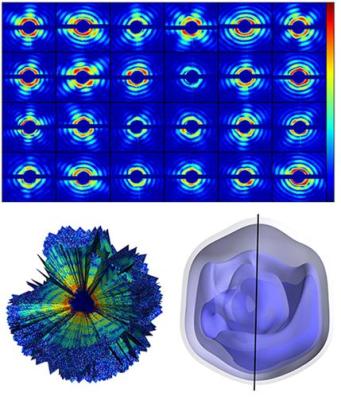 By combining diffraction patterns of single viruses measured with the intense beam from Stanford's X-ray free-electron-laser (XFEL), a team of researchers reconstructed the 3-D structure of the mimivirus. Credit: Tomas Ekeberg et al., Phys. Rev. Lett. 114, 098102 (2015).
By combining diffraction patterns of single viruses measured with the intense beam from Stanford's X-ray free-electron-laser (XFEL), a team of researchers reconstructed the 3-D structure of the mimivirus. Credit: Tomas Ekeberg et al., Phys. Rev. Lett. 114, 098102 (2015).
Solving the structure of single molecules would have profound implications for biology, as many important molecules cannot be crystallized. This is the case for many proteins that interact with biological membranes ("membrane proteins"), which play a role in the immune response, in cellular ion channels and receptors, and are so crucial for life that a very large fraction (20-30%) of the human genome is dedicated to keeping the code for their production.
The authors studied the giant mimivirus, a virus isolated in 1992 from amoebae growing in a water tower and thought to be the cause of certain forms of pneumonia. The team injected single viruses into LCLS' x-ray beam and recorded about 200 diffraction patterns from individual viruses. Since the injected viruses were oriented in unknown directions, the team applied a recently developed optimization algorithm to recover, from the diffraction patterns, the relative orientation of the particles captured by each snapshot. By adding up all contributions they obtained a three-dimensional image of the giant mimivirus.
The work poses an important step towards solving the structures of non-crystallizable biomolecules, demonstrating that it is possible to gather noisy diffraction data from hundreds of individual samples and assemble the data into a complete diffraction pattern, from which the 3D structure can be derived. The technique's spatial resolution (currently 125 nanometer) will need to be improved before biomolecules like proteins, much smaller than the giant mimivirus, can be tackled. However, the method could already image important pathogenic viruses like HIV, influenza and herpes.