Polyaromatic hydrocarbons (PAHs) form an important class of molecules, which can perhaps be regarded as small, discrete graphene species and which play a prominent role in the development of organic electronics. They are also of astrophysical interest as a substantial fraction of the interstellar carbon is believed to be locked up in these very stable molecules. Fundamental understanding of the electronic structure of PAHs and its relation with topological features of the molecules is pursued by these diverse fields.
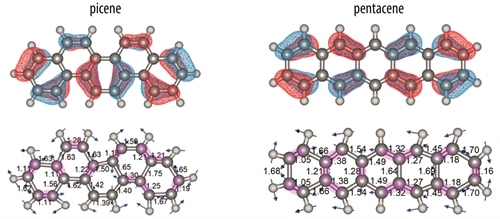 Vibrational mode during one phase of the oscillation (bottom) and molecular orbital representation (top) for the two ions. Anti-nodes in the MO coincide with contracting bonds (purple shading) on the left side of picene and with expanding bonds on the right side. In pentacene, such a clear correlation is not obvious and charge fluxes are largely cancelled. (Credits: Radboud University)
Vibrational mode during one phase of the oscillation (bottom) and molecular orbital representation (top) for the two ions. Anti-nodes in the MO coincide with contracting bonds (purple shading) on the left side of picene and with expanding bonds on the right side. In pentacene, such a clear correlation is not obvious and charge fluxes are largely cancelled. (Credits: Radboud University)
This paper describes an experimental verification of the interaction between electronic and vibrational dynamics in two isomeric PAH species, which differ in their edge topology. Pentacene has a zigzag edge structure, while that of picene is commonly referred to as armchair. Both PAHs are studied in their radical cation form, i.e. an electron has been removed from the highest occupied molecular orbital and the molecules are positively charged. This brings electronically excited states in closer energetic proximity to the ground state and enhances the interaction between them.
The vibrational spectra of the two ions have been recorded using Radboud University’s infrared free-electron laser FELIX coupled to an ion trap mass spectrometer, where the fully isolated, gaseous molecular ions are stored. While the spectra of the two species may appear qualitatively similar, there are substantial differences in the relative intensities of the bands in the various wavelength ranges of the spectra. This paper relates these differences to altered coupling between nuclear and electronic degrees of freedom in the two ions.
The Born-Oppenheimer approximation (BOA) is well known among molecular physicists and physical chemists and constitutes a strict separation between electronic and nuclear motion. While convenient in theories of molecular physics, many manifestations of its limitations are also known and the differences in the relative intensities in the vibrational spectra of pentacene and picene cations present another example.
Qualitatively, one can imagine a vibrational normal mode as an oscillatory motion of the atoms within a molecule, whereby certain bonds contract while others expand; during the opposite phase of this oscillatory motion, contraction and expansion are reversed. While ignored in the BOA formalism, it is straightforward to realize that the overlap between valence atomic orbitals increases during the contraction phase of a vibrational period, while it decreases when the bond expands. The energies of bonding and anti-bonding molecular orbitals thus ought to oscillate as the molecule vibrates.
Each molecular orbital (wave function) contains nodes and anti-nodes located roughly over the bonds in the molecule. For specific combinations of molecular orbitals and vibrational normal modes, it may occur that on one side of the molecule, all anti-nodes in the wave function coincide with contracting bonds during one phase of the vibrational oscillation; imposed by symmetry, the anti-nodes on the other side of the molecule then coincide with expanding bonds. It appears as if the molecule becomes more bonding on one side, while less bonding on the other side. A net electron density shift to the more bonding side occurs. Half a vibrational period later, the situation reverses: now the other side of the molecule is more bonding and electron density shifts to that side. This vibrationally induced oscillating electron flux generates an additional dipole derivative that enhances the intensity of the particular vibrational mode.
Whether or not there are combinations of molecular orbitals and vibrational normal modes that coincide such that a dynamic electron flux is generated is dependent on the topology and the symmetry of the molecule. Here, we experimentally show that such an enhancement occurs for picene, but that the effect largely cancels in pentacene, where some of anti-nodes on one side of the molecule coincide with contracting bond while others coincide with expanding bonds. Based on symmetry considerations, this is inferred to be typical for zigzag versus armchair polyaromatic systems.
This project was financed by NWO (the Netherlands Organisation for Scientific Research) and FOM (the Dutch Foundation for Fundamental Research on Matter).