|
Scientists at the SFI Nanoscience Laboratory at Trinity College, Dublin, Ireland, and the University of North Carolina at Chapel Hill, have used Accelrys' CASTEP to study possible bonding geometries of 1,3-cyclohexadiene (1,3-CHD) on a silicon(100) surface.
These reactions are of particular interest for the integration of organic molecules and molecular thin films into existing silicon-based technology. Comparison with the product distribution obtained by Scanning Tunneling Microscope (STM) experiments shows a kinetically controlled reaction.
The results will aid the development of smaller, faster electronic devices.
Existing Silicon-Based Technology
Existing silicon-based technology has an ever-increasing demand for the manufacture of smaller and faster devices. Recently, the ability to integrate organic molecules and molecular thin films on silicon surfaces has been dramatically improved by experimental observations that small organic molecules can attach to Si(100) surfaces via the Si-Si dimer bond. If organics can form molecular building blocks on Si(100), a detailed understanding of the reaction mechanism and the factors that control the attachment of the molecule on the surface is essential.
Using CASTEP to Investigate Different Bonding Geometries of 1,3-Cyclohexadiene
In this study, the plane-wave based Density Functional Theory (DFT) program CASTEP was used to investigate the different possible binding geometries of 1,3-cyclohexadiene (1,3-CHD) on a silicon(100) surface. The results were then compared to recent STM experiments.
Results
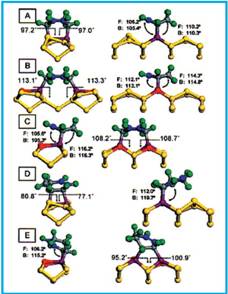
All possible adducts were fully optimized and the results were analyzed in terms of total energies and the detailed geometries. The obtained optimized adducts are shown in Fig. 1.
|
|
|
Figure 1. CASTEP-optimized structures for different binding modes of 1,3-CHD on 6 Si(100).
|
The Most Likely Bonding Mechanisms for 1,3-Cyclohexadiene on Silicon
The researchers found that the products resulting from [4+2] cycloaddition (structures A-C in Fig.1) are energetically favored over [2+2] cycloaddition, in agreement with earlier calculations. Of the [4+2] adducts, structure A in Fig. 1 is favored. However, when allowance of surface-dangling bonds is made, structures B and C straddle the inter-dimer product A, being about 3 kcal/mol higher or lower in energy, respectively. This energy ordering can be rationalized on the basis of ring strain arguments.
Experimental observations show that, in addition to the kinetically favored [4+2] adduct, there is a significant (about 20%) population of the [2+2] adducts, which is inconsistent with transition-state calculations that predicted an absence of an energy barrier for the [4+2] addition, where as a barrier was found for the [2+2] cycloaddition. Whether there are dynamical bottlenecks associated with specific geometries required for reactions remains to be established. It is clear that the reactivity of 1,3-CHD on Si(100) is high. However, the reaction is not indiscriminate, since products that are accessible (structures on the right hand side of Fig. 1) are explicitly excluded from the experimentally observed surface population
|