A head-to-head benchmark shows why the newest algorithm is not always the best choice and how sequencing context can determine which methylation caller researchers should trust.
Study: Comprehensive benchmarking of tools for nanopore-based detection of DNA methylation. Image Credit: AI-generated using ChatGPT/OpenAI
Nanopore sequencing can detect DNA base modifications directly from native DNA, but accurately identifying these chemical marks remains challenging. A recent paper, published online as an 'Article in Press' in Nature Communications, systematically benchmarked software tools for detecting DNA modifications from nanopore sequencing data.
Researchers found that specific newer models offered the best overall performance for non-CpG 5-methylcytosine (5mC), meaning methylated cytosines outside cytosine-guanine, or CpG, sites, 6-methyladenine (6mA), and 4-methylcytosine (4mC), whereas the older Dorado v4r1 model and RockFish remained the most reliable for CpG methylation profiling. These findings provide practical guidance for selecting computational tools for epigenetic research and nanopore-based genomic analysis.
The Role of Nanopore Sequencing
DNA methylation is an important epigenetic modification that contributes to the regulation of gene expression, genome stability, and cellular development. Traditional methods for detecting these modifications, such as bisulfite sequencing, require chemical treatment that can damage DNA, introduce amplification bias, and complicate mapping in complex genomic regions.
In this context, nanopore sequencing provides a direct alternative by analyzing native DNA without chemical conversion. As individual DNA molecules pass through a bioengineered nanopore, changes in electrical current reveal the presence of modified bases.
Oxford Nanopore's R10 flow cells use a more stable, bioengineered nanopore with a longer sensing region, improving the resolution of homopolymer sequences and overall sequencing accuracy. Neural network-based algorithms then interpret these electrical signals to identify DNA base modifications alongside the nucleotide sequence. As sequencing accuracy has improved, the main challenge has shifted from signal acquisition to the computational interpretation of these complex electrical signals.
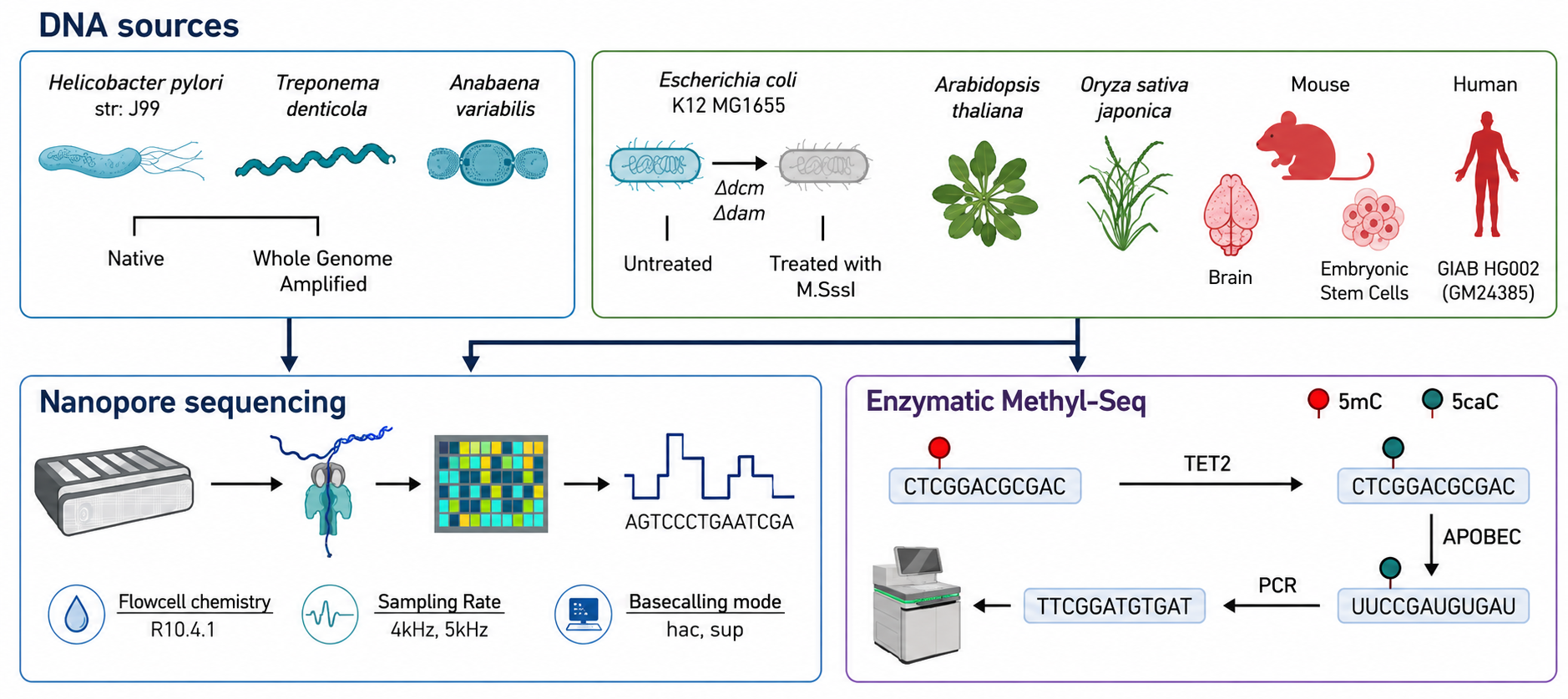
Sources of DNA and whole genome sequencing workflow used in this study. Genomic DNA from bacterial/plant/mammalian samples was sequenced on the R10.4.1 flowcells. A subset of samples (right box on the top) were also subject to Enzymatic Methyl-Seq (EMSeq) as ground truth for 5-methylcytosine (5mC). Image adapted from fig 1a. Kulkarni, O., et al. (2026). Comprehensive benchmarking of tools for nanopore-based detection of DNA methylation. Nat Commun. DOI: 10.1038/s41467-026-75183-6 using ChatGPT/OpenAI.
Framework for Benchmarking Methylation Tools
To evaluate current computational methods for nanopore methylation analysis, researchers benchmarked widely used software using whole-genome sequencing data from diverse biological sources. These data were generated from five bacterial species, two plant species, and mammalian samples, including mouse whole-brain tissue, mouse embryonic stem cells, and the human HG002 cell line. This diverse dataset captured a broad range of DNA modifications, from the common methylation patterns found in mammals to less frequent modifications present in plants and bacteria.
The datasets included sequencing data generated using Oxford Nanopore R10.4.1 flow cells at sampling rates of 4 kHz and 5 kHz. Enzymatic Methyl-seq provided the 5mC reference data for the plant and mouse samples and E. coli, while the human HG002 data were compared with a publicly available whole-genome bisulfite sequencing dataset. Public Pacific Biosciences data provided 6mA and 4mC comparisons for three bacterial species, while REBASE motif information was used for the other two species.
The raw nanopore signals were processed with DeepBAM, DeepMod2, DeepPlant, f5C, RockFish, and multiple versions of the Dorado models. The benchmark compared each model under different operating modes, including high-accuracy and super-accuracy settings. The study also evaluated detection accuracy, false-positive rates, processing speed, memory use, and the effects of sequencing depth, read quality, and neighboring DNA modifications by comparing nanopore predictions with the reference datasets.
Performance and Algorithmic Limitations
The benchmark showed clear differences in performance across computational models. For standard CpG methylation, Dorado v4r1 and RockFish achieved the highest accuracy and agreement with the reference datasets. Although newer Dorado models showed lower accuracy in routine CpG methylation profiling due to higher false-negative rates, Dorado v5r3 performed best overall for non-CpG 5mC and 4mC, while Dorado v5r1 was preferred overall for 6mA. The latest v5.2 models reduced some false-positive effects from neighboring modifications but were not consistently superior because several had poorer recall.
The analysis also identified important limitations shared by many algorithms. Because the electrical signal measured by a nanopore reflects multiple neighboring bases, nearby DNA modifications could increase false-positive or false-negative calls depending on the modification, sequence context, distance, and model.
DeepPlant performed well for non-CpG methylation in plant data when individual DNA reads were evaluated, but performed poorly on mammalian datasets, indicating a strong species-specific training bias. Dorado v5r3 generally provided better performance when methylation estimates were aggregated at individual genomic sites. Computational performance also varied considerably. Dorado provided the highest overall throughput in the bacterial benchmarks while using 12–14 GB of memory, whereas DeepPlant required an average of more than 84 GB. However, f5C outperformed the super-accuracy Dorado 5mCG model in both speed and memory use.
The study also found that correlation in rice CpG analyses generally plateaued after a median coverage of about 20×, although error continued to decrease at higher coverage. The authors therefore recommended at least 20× median coverage. In three bacterial 6mA datasets, filtering reads below a Phred quality score of 20 reduced quality-dependent undercalling and produced more consistent methylation estimates.
Study Limitations
The study used Enzymatic Methyl-seq as reference data despite its potential biases, did not benchmark 5-hydroxymethylcytosine, and evaluated 4mC across relatively few sequence contexts. It also relied on REBASE rather than on direct Pacific Biosciences data for two bacterial species, and some analyses were limited to chromosome 1 or required large datasets to be split into smaller computational batches.
Implications for Epigenetic Research
This research offers practical guidance for selecting computational tools for nanopore-based epigenetic analysis. By matching algorithms to specific DNA modifications, scientists can increase the accuracy of methylation profiling while reducing analytical errors. These findings are particularly valuable for plant genomics, where accurate detection of non-CpG methylation can support research into development, stress responses, and transposon silencing.
More generally, reliable detection of DNA methylation from native DNA supports the use of nanopore sequencing in epigenetic studies. However, this benchmarking study did not evaluate clinical samples, diagnostic accuracy, precision medicine applications, crop traits, or disease biomarkers. As computational methods advance, nanopore sequencing may become a more dependable tool for studying disease-associated epigenetic changes and other relevant biomarkers, but these potential applications require separate validation.
Future Directions in Nanopore Sequencing
In summary, this benchmarking study provides a comprehensive assessment of current nanopore methylation analysis tools, highlighting both their strengths and their limitations. While advances in nanopore sequencing hardware have improved data quality, accurate interpretation of DNA modifications still depends on robust computational methods.
Future work should focus on further developing algorithms that better account for the influence of neighboring DNA modifications while maintaining high accuracy and computational efficiency. The open-access datasets and benchmarking framework established by the researchers provide a valuable resource for refining nanopore methylation analysis and supporting future advances in epigenetics and genomics.
Disclaimer: The views expressed here are those of the author expressed in their private capacity and do not necessarily represent the views of AZoM.com Limited T/A AZoNetwork the owner and operator of this website. This disclaimer forms part of the Terms and conditions of use of this website.