With the passage of a molecule through the labyrinth of a chemical system being so critical to catalysis and other important chemical processes, computer simulations are frequently used to model potential molecule/labyrinth interactions.
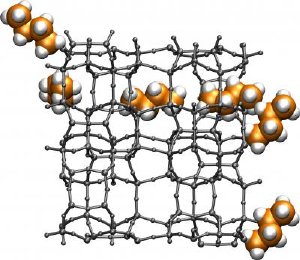 This figure shows a molecular worm representing a butane molecule as it navigates through the chemical labyrinth of a typical alkane-cracking zeolite. The alogorithm was used to compute the shortest path for the butane molecule to traverse one unit of the periodic zeolite structure. Credit: Image courtesy of Maciej Haranczyk, Berkeley Lab
This figure shows a molecular worm representing a butane molecule as it navigates through the chemical labyrinth of a typical alkane-cracking zeolite. The alogorithm was used to compute the shortest path for the butane molecule to traverse one unit of the periodic zeolite structure. Credit: Image courtesy of Maciej Haranczyk, Berkeley Lab
In the past, such simulations have been expensive and time-consuming to carry out, but now researchers with the Lawrence Berkeley National Laboratory (Berkeley Lab) have developed a new algorithm that should make future simulations easier and faster to compute, and yield much more accurate results.
"Currently the major limiting factor in running molecular simulations for a large number of structures before they can be screened for useful materials is the need to visually analyze the structures to set up successful simulations," says Maciej Haranczyk, a computational chemist and a 2008 Glenn T. Seaborg Fellow in Berkeley Lab's Computational Research Division. "With our approach, such structural analysis can be done automatically, which speeds up the whole process of material screening."
Haranczyk is co-author of a paper that appears in the Proceedings of the National Academy of Sciences entitled: "Navigating molecular worms inside chemical labyrinths." The other author of this paper is James Sethian, who heads the Mathematics Group of Berkeley Lab's Computational Research Division, and is also a professor in the Mathematics Department of the University of California, Berkeley.
A key to the success of this new algorithm was its departure from the traditional treatment of molecules as hard spheres with fixed radii. Instead, Haranczyk and Sethian constructed "molecular worms" from blocks connected by flexible links. These molecular worms provide a more realistic depiction of a molecule's geometry, thereby providing a more accurate picture of how that molecule will navigate through a given chemical labyrinth, as Sethian explains.
"In practice, most molecules of interest, even the simplest solvents or gases, rarely have a spherical shape, and treating molecules as such may lead to errors," he says. "Our molecular worms are able to change shape during the traversing of a chemical labyrinth, which allows them to reach areas not accessible to either a single large spherical probe or a rigid real-shape probe. This significantly extends the range of probes and structures that can be efficiently examined."
As a molecule navigates through a chemical system, its access to a particular site or place within that system determines the extent to which catalysis and other chemical reactions may occur. Many of these critical sites are either buried in clefts, pockets or hidden cavities, or else represent channel systems. The accessible volume of a chemical system – the free volume available to a penetrating molecule - is also critical to the system's physical properties, including diffusion, viscosity and electrical conductivity. Predicting whether a molecule will be able to traverse through a given chemical labyrinth is the first question that a simulation must answer, followed by identifying the shortest transverse route, finding the largest probe that can transverse though the system, and calculating accessible volume.
"The required calculations become quite expensive as one needs to include interactions of all the atoms of the penetrating molecule with all the atoms in the labyrinth, and this procedure has to be repeated at every step of the simulation," Haranczyk says. "Additionally, in molecular dynamics only one trajectory per molecule is investigated. Since a penetrating molecule can be bouncing off the walls of a system before it finds a way out, mapping accessible volume in a chemical labyrinth may require the running of a very long simulation to actually see the molecule moving through."
Haranczyk, looking to automate the process by which the void spaces of porous materials are analyzed, had an idea for a probe that would walk through the inside a material and map it. Sethian had been working on mathematic techniques that can be used in robotic navigations and path planning, as well as a host of algorithms for computing geometries in complex settings.
"What's exciting here is to bring together two disparate worlds to build a new technology" says Sethian. The two scientists pooled their expertise to develop the molecular worm algorithm, which they first tested on a zeolite material. Zeolites are microporous minerals that have been widely used since the late 1950s as chemical catalysts, membranes for separations, and water softeners. They are especially useful as alkane-cracking catalysts in oil refinement.
"There are 190 zeolite structures known to exist today, but they constitute only a very small fraction of the 2.5 million structures that are feasible on theoretical grounds," Haranczyk says. "The development of a database of hypothetical zeolite structures has long been regarded as an important step toward designer catalysts as it could, in principle, be screened for zeolites of any property. However, brute-force screening of all possible zeolite structures through molecular dynamics characterization is computationally infeasible, hence the need for rapid triaging based on an initial analysis of various properties."
The successful testing of the molecular worm algorithm on a typical alkane-cracking zeolite opens an immediate door to its use in screening for new zeolites as well as a wide variety of other porous materials. The algorithm should also prove valuable in the search for materials that can capture carbon emissions before they enter the atmosphere. With further refinements, it could also one day be applied to proteins, especially enzymes.
"Being at the frontier of science and solving a very complex problem that has not been addressed before is always very exciting," Haranczyk says.